Un equipo de investigadores de la UNLP, junto a científicos de la Universidad de Rennes, Francia, estudian defectos congénitos asociados a una serie de síndromes conocidos como holoprosencefalia (HPE). Se trata de una malformación que afecta severamente el rostro y el cerebro, y que se manifiesta por una división incompleta del prosencéfalo, que es la porción anterior del cerebro durante la fase de desarrollo del embrión.
La HPE es una enfermedad de las denominadas raras. Según estadísticas, una de cada 250 concepciones presenta HPE, y una parte no prospera y muere en el caso de las formas graves. Se estima que uno de cada 200 abortos espontáneos en humanos son causados por estas anomalías. De los casos que no son graves y llegan a nacer, la frecuencia de casos de HPE es de 1 por cada 10.000 nacimientos.
“Nuestra investigación tiene como objetivo describir los mecanismos de una anormalidad en el desarrollo temprano del cerebro, denominada holoprosencefalia (HPE)”, explicó el Doctor Luis Diambra, quien está a cargo del Laboratorio de Biología de Sistemas del Centro Regional de Estudios Genómicos (CREG), de la Facultad de Ciencias Exactas UNLP e investigador del CONICET, y es integrante del equipo dirigido por la Doctora Marie De Tayrac, en la Universidad Rennes.
En 1997 se creó en Francia un grupo de 2000 pacientes con HPE y sus padres. En los estudios previos identificaron mutaciones en unos 15 genes que están implicados en el 30% de los casos. Un efecto común de estas mutaciones es la disminución de la señal de Sonic Hedgehog (SHH) que conduce a un defecto en la región rostroventral (la parte delantera del rostro) del embrión y la HPE.
Se ha establecido que la HPE es una enfermedad genética compleja, en la que las malformaciones son el resultado de múltiples relaciones entre diferentes genes. Para estudiarla, los genetistas están trabajando con bioinformáticos y biólogos del desarrollo para identificar nuevas mutaciones asociadas a la severidad de HPE
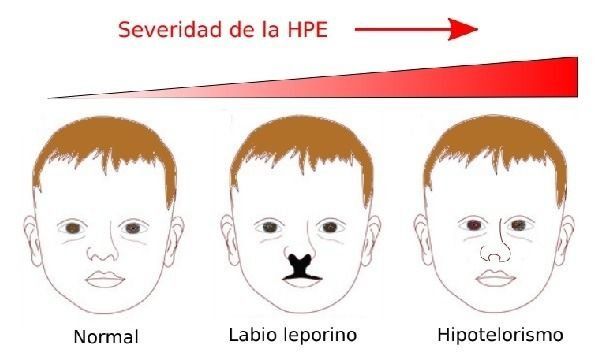
Las formas menos graves de HPE son: un diente incisivo único central, obstrucciones nasales, displasia retiniana, labio leporino, hendidura en el paladar, o disminución de la distancia entre ojos (hipotelorismo). Los casos más graves, como ciclopía y proboscis, suelen ser fatales.
“Más allá de identificar nuevos genes candidatos para HPE, estamos ayudando a comprender los mecanismos moleculares involucrados en los casos menos graves de HPE. En este sentido lo interesante y novedoso de nuestro trabajo es que las mutaciones en el gen SHH asociadas con los casos leves de HPE son del tipo mutaciones sinónimas, también llamadas ‘silenciosas’. Son aquellas que cambian el codón original por otro que determina el mismo aminoácido”, señaló el Dr. Biambra.

En esa línea, agregó: “La pregunta que se viene realizando la ciencia es ¿por qué entonces una mutación sinónima, que no cambia la secuencia de la proteína, puede tener alguna consecuencia? Al parecer, estas mutaciones sinónimas no son tan inocuas como se pensaba. Observamos que los portadores de estas mutaciones tienen menos cantidad de la proteína SHH”.
Esta investigación fue recientemente publicada en la importante revista científica Brain de la Universidad de Oxford, en el siguiente link: https://academic.oup.com/brain/advance-article-abstract/doi/10.1093/brain/awaa152/5857793
El descubrimiento no ofrece un tratamiento para evitar malformaciones congénitas, para restaurar la apariencia y la función normal de malformaciones, como el labio leporino, es usual las cirugías correctivas en recién nacidos. Sin embargo, este hallazgo científico contribuye a las herramientas para determinar los riesgos de tener de defectos congénitos cuando existen antecedentes familiares. Por otro lado, el estudio confirma la hipótesis que las mutaciones silenciosas pueden no serlo tanto como se pensaba.